



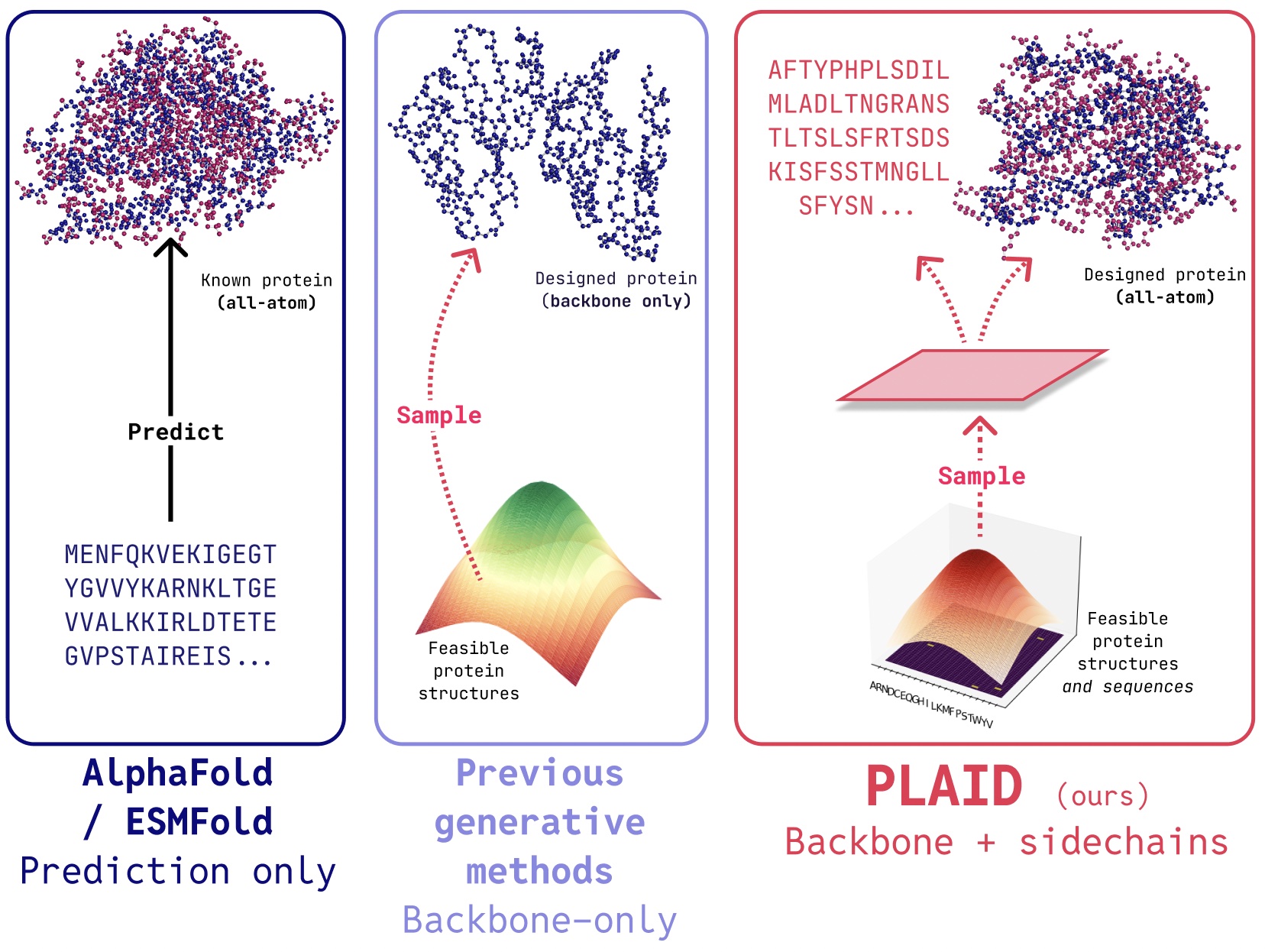
PLAID is a multimodal generative mannequin that concurrently generates protein 1D sequence and 3D construction, by studying the latent house of protein folding fashions.
The awarding of the 2024 Nobel Prize to AlphaFold2 marks an vital second of recognition for the of AI position in biology. What comes subsequent after protein folding?
In PLAID, we develop a way that learns to pattern from the latent house of protein folding fashions to generate new proteins. It may settle for compositional operate and organism prompts, and will be educated on sequence databases, that are 2-4 orders of magnitude bigger than construction databases. Not like many earlier protein construction generative fashions, PLAID addresses the multimodal co-generation downside setting: concurrently producing each discrete sequence and steady all-atom structural coordinates.
From construction prediction to real-world drug design
Although current works exhibit promise for the power of diffusion fashions to generate proteins, there nonetheless exist limitations of earlier fashions that make them impractical for real-world functions, resembling:
- All-atom era: Many present generative fashions solely produce the spine atoms. To supply the all-atom construction and place the sidechain atoms, we have to know the sequence. This creates a multimodal era downside that requires simultaneous era of discrete and steady modalities.
- Organism specificity: Proteins biologics supposed for human use should be humanized, to keep away from being destroyed by the human immune system.
- Management specification: Drug discovery and placing it into the fingers of sufferers is a fancy course of. How can we specify these complicated constraints? For instance, even after the biology is tackled, you may determine that tablets are simpler to move than vials, including a brand new constraint on soluability.
Producing “helpful” proteins
Merely producing proteins is just not as helpful as controlling the era to get helpful proteins. What may an interface for this appear to be?

For inspiration, let’s think about how we would management picture era by way of compositional textual prompts (instance from Liu et al., 2022).
In PLAID, we mirror this interface for management specification. The final word purpose is to manage era completely by way of a textual interface, however right here we think about compositional constraints for 2 axes as a proof-of-concept: operate and organism:
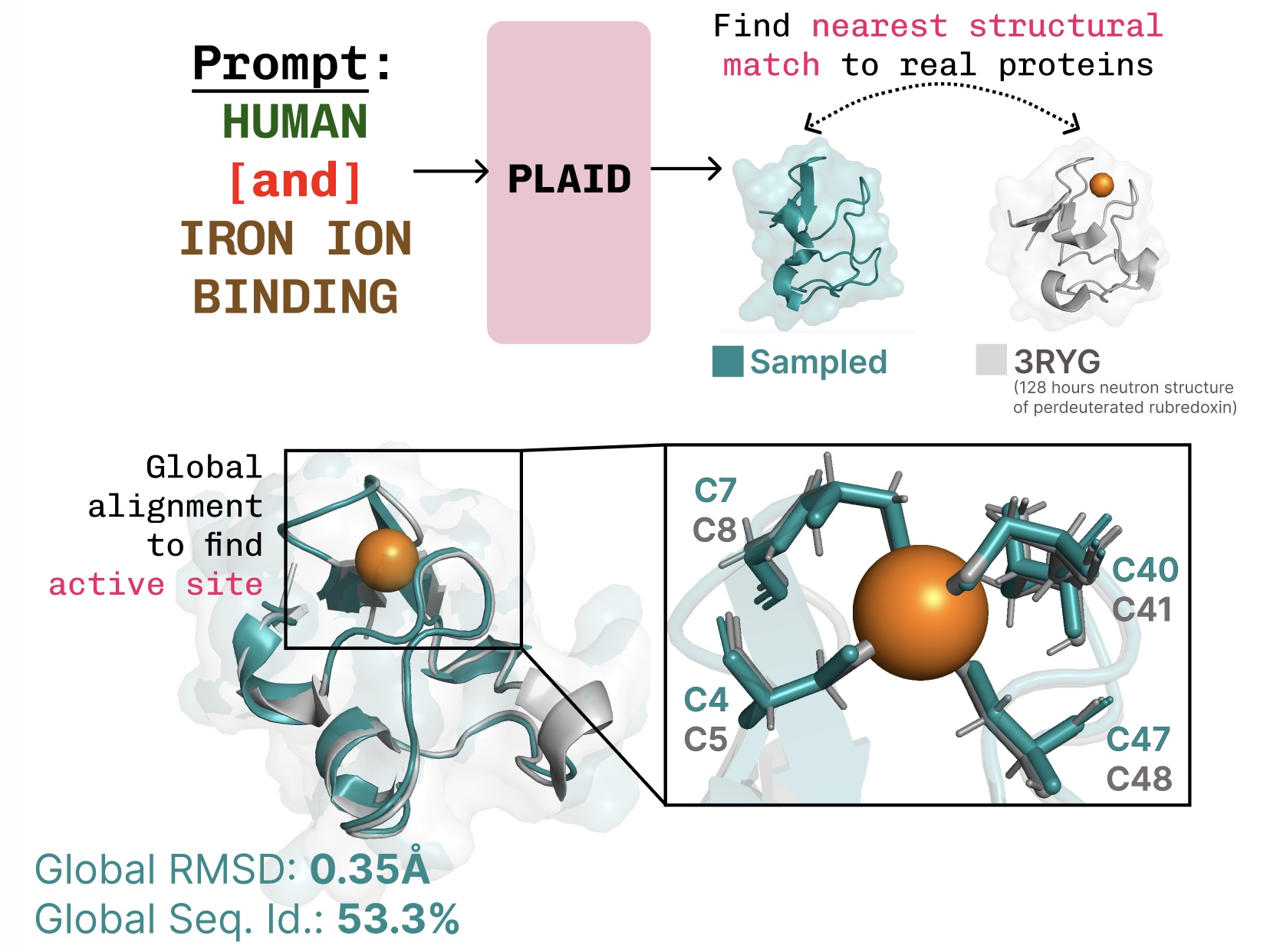
Studying the function-structure-sequence connection. PLAID learns the tetrahedral cysteine-Fe2+/Fe3+ coordination sample typically present in metalloproteins, whereas sustaining excessive sequence-level variety.
Coaching utilizing sequence-only coaching information
One other vital side of the PLAID mannequin is that we solely require sequences to coach the generative mannequin! Generative fashions be taught the information distribution outlined by its coaching information, and sequence databases are significantly bigger than structural ones, since sequences are less expensive to acquire than experimental construction.
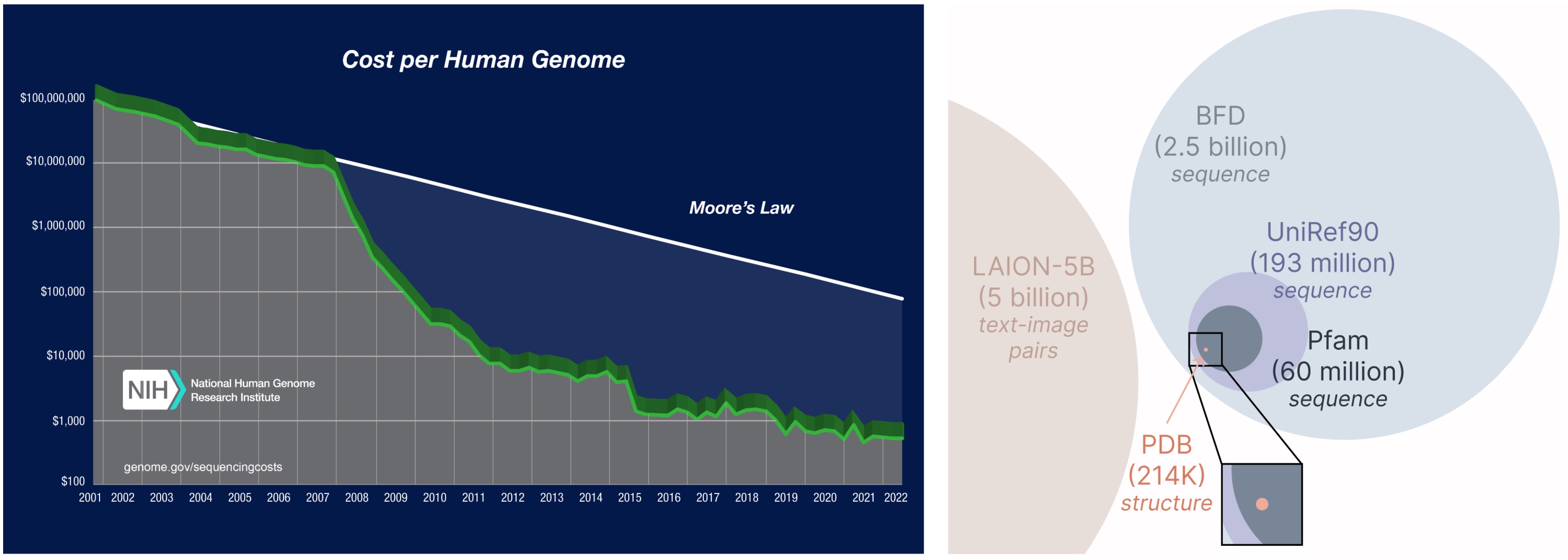
Studying from a bigger and broader database. The price of acquiring protein sequences is way decrease than experimentally characterizing construction, and sequence databases are 2-4 orders of magnitude bigger than structural ones.
How does it work?
The rationale that we’re capable of prepare the generative mannequin to generate construction by solely utilizing sequence information is by studying a diffusion mannequin over the latent house of a protein folding mannequin. Then, throughout inference, after sampling from this latent house of legitimate proteins, we are able to take frozen weights from the protein folding mannequin to decode construction. Right here, we use ESMFold, a successor to the AlphaFold2 mannequin which replaces a retrieval step with a protein language mannequin.
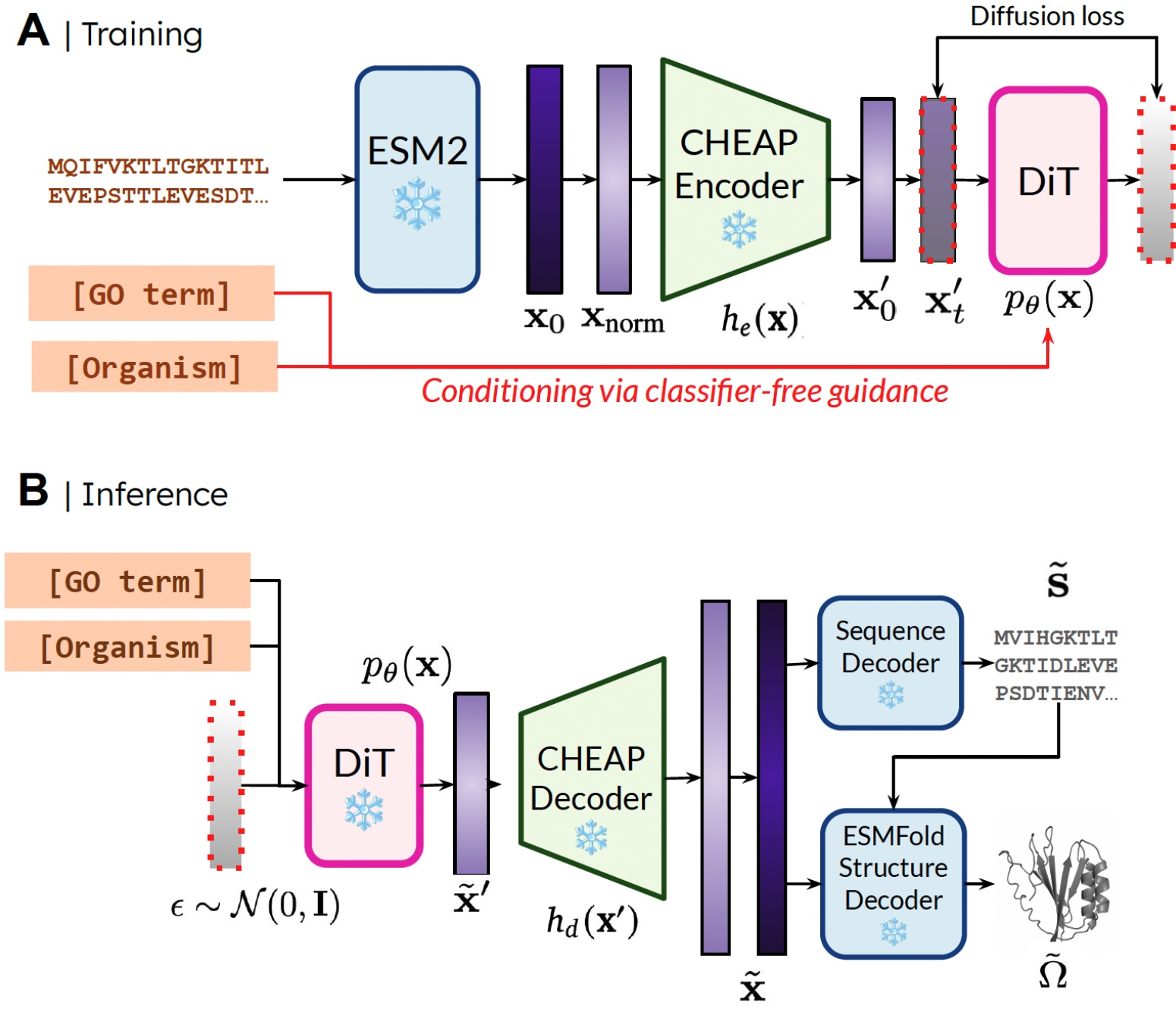
Our methodology. Throughout coaching, solely sequences are wanted to acquire the embedding; throughout inference, we are able to decode sequence and construction from the sampled embedding. ❄️ denotes frozen weights.
On this means, we are able to use structural understanding info within the weights of pretrained protein folding fashions for the protein design activity. That is analogous to how vision-language-action (VLA) fashions in robotics make use of priors contained in vision-language fashions (VLMs) educated on internet-scale information to produce notion and reasoning and understanding info.
Compressing the latent house of protein folding fashions
A small wrinkle with straight making use of this methodology is that the latent house of ESMFold – certainly, the latent house of many transformer-based fashions – requires quite a lot of regularization. This house can be very massive, so studying this embedding finally ends up mapping to high-resolution picture synthesis.
To deal with this, we additionally suggest CHEAP (Compressed Hourglass Embedding Diversifications of Proteins), the place we be taught a compression mannequin for the joint embedding of protein sequence and construction.
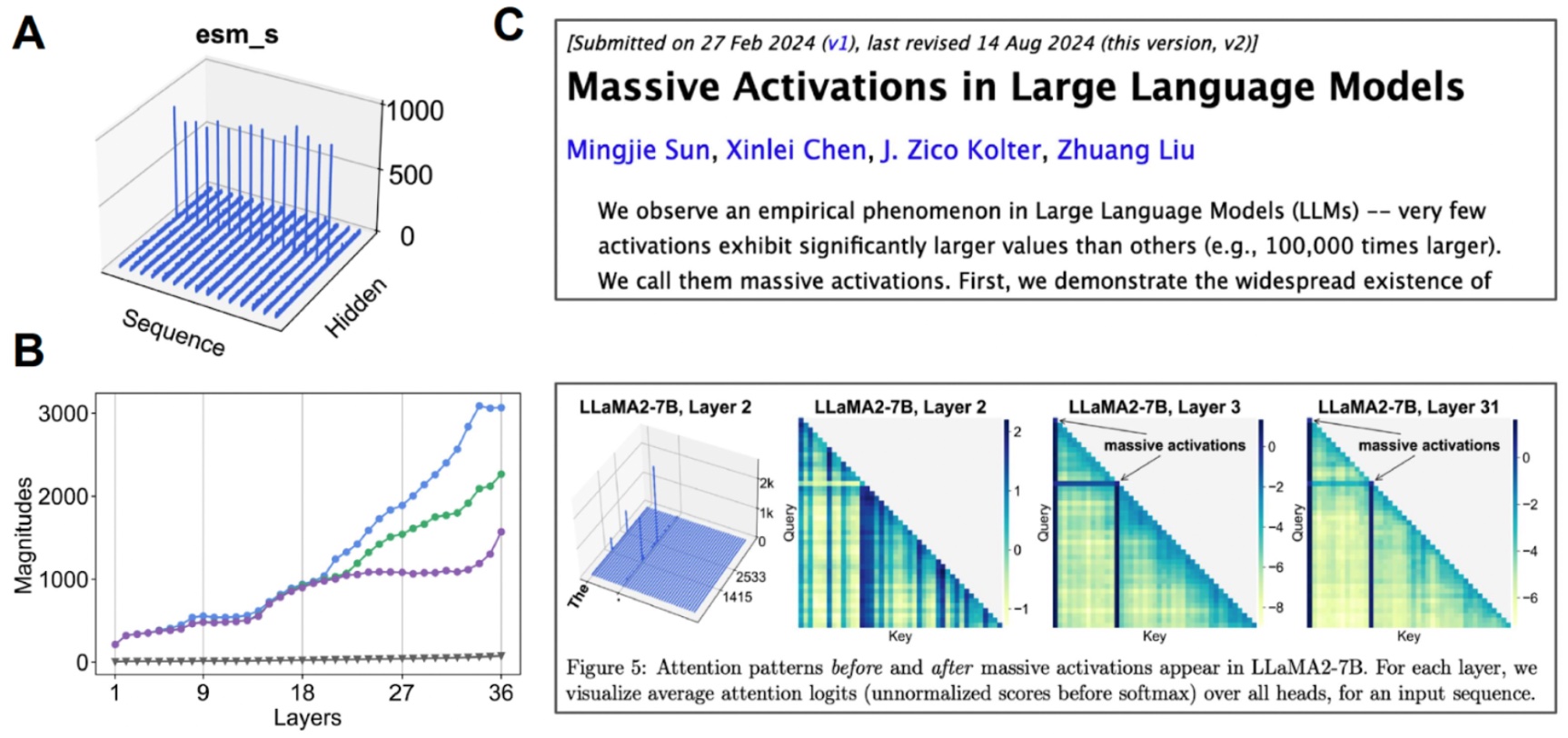
Investigating the latent house. (A) After we visualize the imply worth for every channel, some channels exhibit “huge activations”. (B) If we begin analyzing the top-3 activations in comparison with the median worth (grey), we discover that this occurs over many layers. (C) Huge activations have additionally been noticed for different transformer-based fashions.
We discover that this latent house is definitely extremely compressible. By doing a little bit of mechanistic interpretability to higher perceive the bottom mannequin that we’re working with, we have been capable of create an all-atom protein generative mannequin.
What’s subsequent?
Although we study the case of protein sequence and construction era on this work, we are able to adapt this methodology to carry out multi-modal era for any modalities the place there’s a predictor from a extra plentiful modality to a much less plentiful one. As sequence-to-structure predictors for proteins are starting to sort out more and more complicated programs (e.g. AlphaFold3 can be capable of predict proteins in complicated with nucleic acids and molecular ligands), it’s simple to think about performing multimodal era over extra complicated programs utilizing the identical methodology.
If you’re focused on collaborating to increase our methodology, or to check our methodology within the wet-lab, please attain out!
Additional hyperlinks
For those who’ve discovered our papers helpful in your analysis, please think about using the next BibTeX for PLAID and CHEAP:
@article{lu2024generating,
title={Producing All-Atom Protein Construction from Sequence-Solely Coaching Information},
writer={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Steady Embedding Compressions of Protein Sequence and Construction},
writer={Lu, Amy X and Yan, Wilson and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
You can even checkout our preprints (PLAID, CHEAP) and codebases (PLAID, CHEAP).
Some bonus protein era enjoyable!
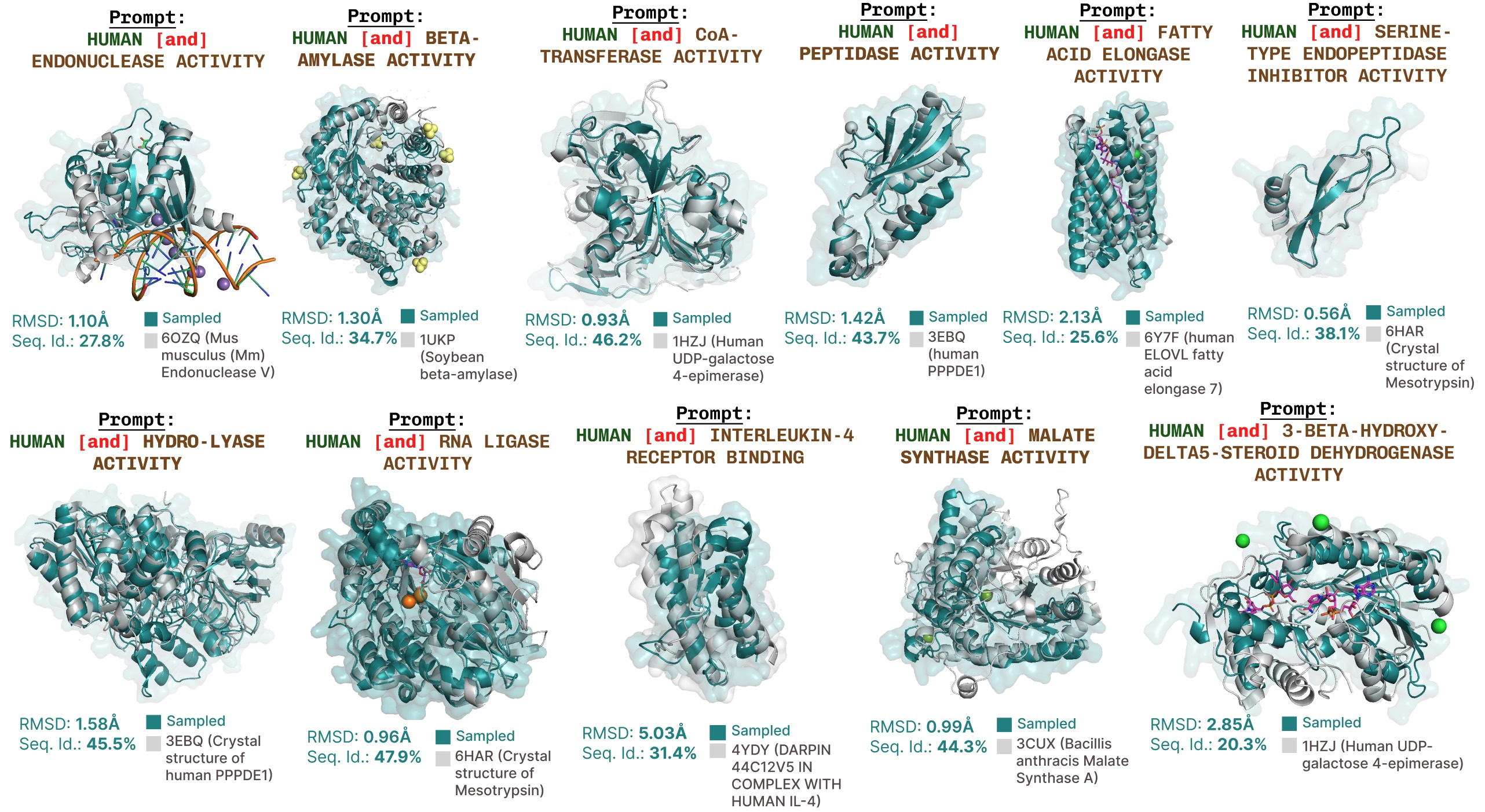
Further function-prompted generations with PLAID.
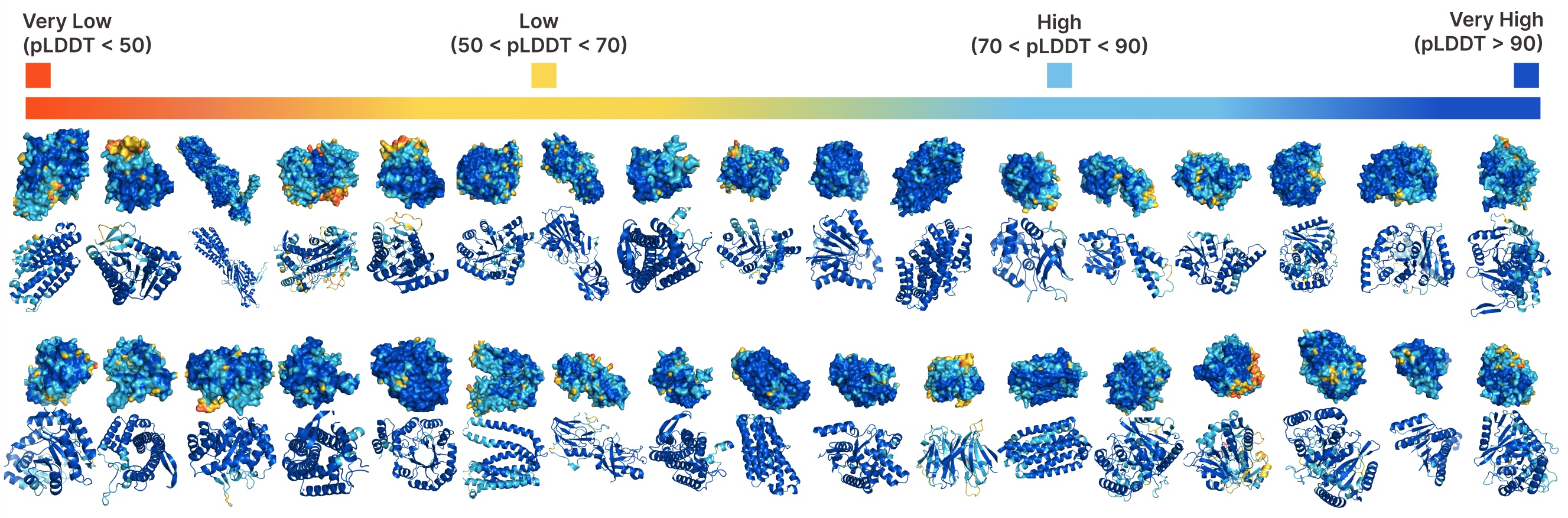
Unconditional era with PLAID.
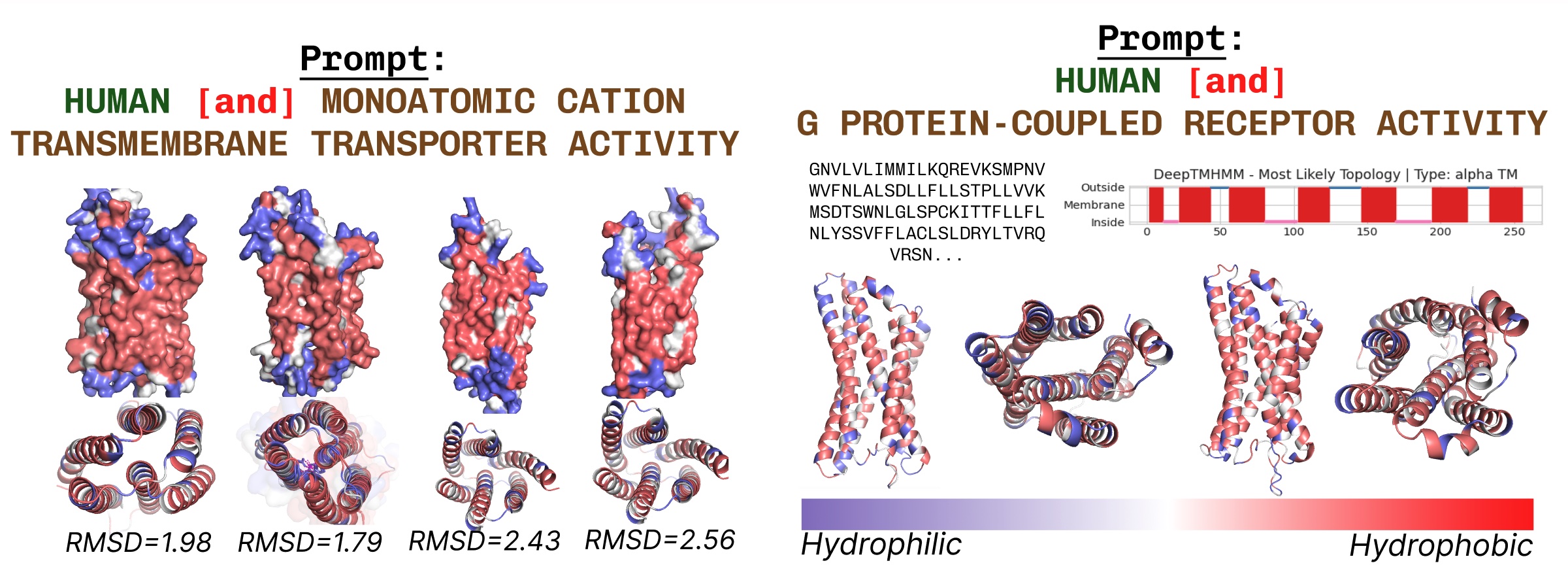
Transmembrane proteins have hydrophobic residues on the core, the place it’s embedded inside the fatty acid layer. These are constantly noticed when prompting PLAID with transmembrane protein key phrases.
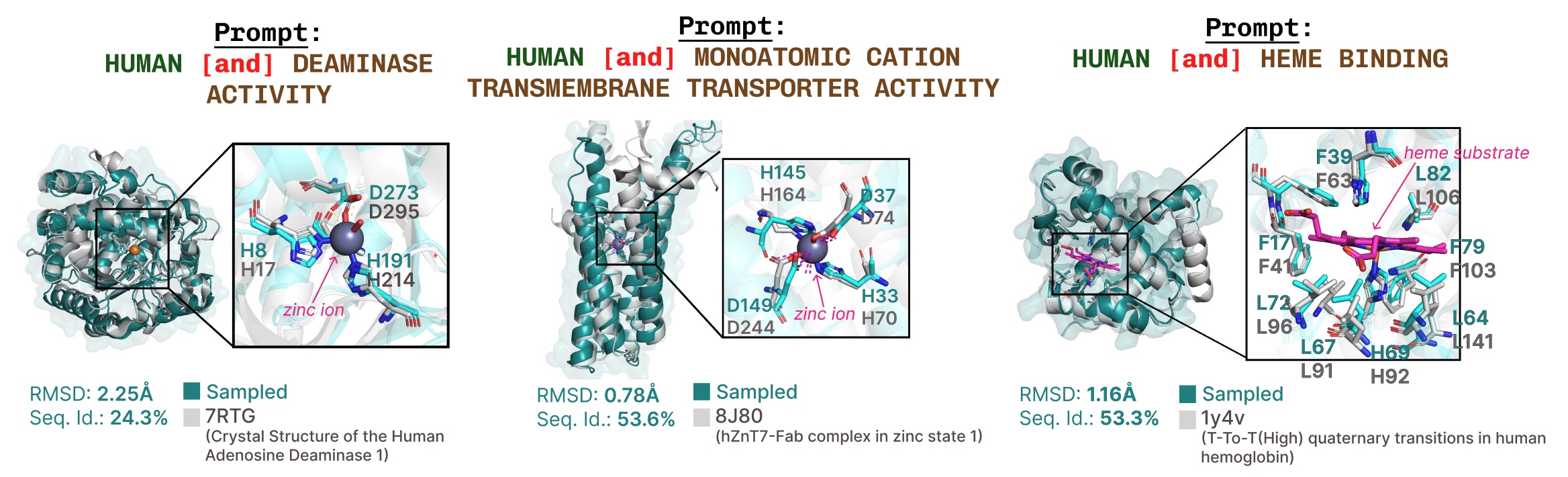
Further examples of lively web site recapitulation based mostly on operate key phrase prompting.
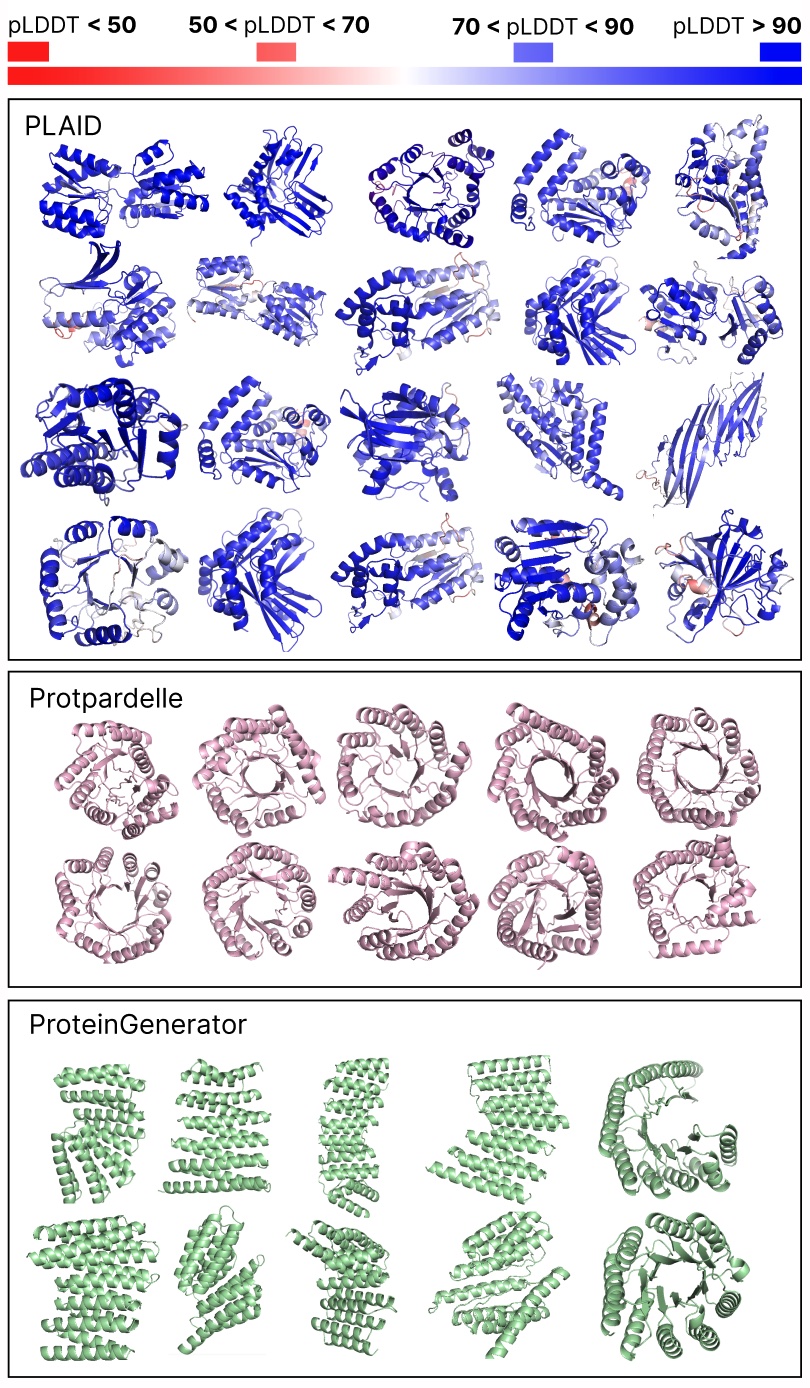
Evaluating samples between PLAID and all-atom baselines. PLAID samples have higher variety and captures the beta-strand sample that has been tougher for protein generative fashions to be taught.
Acknowledgements
Due to Nathan Frey for detailed suggestions on this text, and to co-authors throughout BAIR, Genentech, Microsoft Analysis, and New York College: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin Okay. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, and Nathan C. Frey.
PLAID is a multimodal generative mannequin that concurrently generates protein 1D sequence and 3D construction, by studying the latent house of protein folding fashions.
The awarding of the 2024 Nobel Prize to AlphaFold2 marks an vital second of recognition for the of AI position in biology. What comes subsequent after protein folding?
In PLAID, we develop a way that learns to pattern from the latent house of protein folding fashions to generate new proteins. It may settle for compositional operate and organism prompts, and will be educated on sequence databases, that are 2-4 orders of magnitude bigger than construction databases. Not like many earlier protein construction generative fashions, PLAID addresses the multimodal co-generation downside setting: concurrently producing each discrete sequence and steady all-atom structural coordinates.
From construction prediction to real-world drug design
Although current works exhibit promise for the power of diffusion fashions to generate proteins, there nonetheless exist limitations of earlier fashions that make them impractical for real-world functions, resembling:
- All-atom era: Many present generative fashions solely produce the spine atoms. To supply the all-atom construction and place the sidechain atoms, we have to know the sequence. This creates a multimodal era downside that requires simultaneous era of discrete and steady modalities.
- Organism specificity: Proteins biologics supposed for human use should be humanized, to keep away from being destroyed by the human immune system.
- Management specification: Drug discovery and placing it into the fingers of sufferers is a fancy course of. How can we specify these complicated constraints? For instance, even after the biology is tackled, you may determine that tablets are simpler to move than vials, including a brand new constraint on soluability.
Producing “helpful” proteins
Merely producing proteins is just not as helpful as controlling the era to get helpful proteins. What may an interface for this appear to be?
For inspiration, let’s think about how we would management picture era by way of compositional textual prompts (instance from Liu et al., 2022).
In PLAID, we mirror this interface for management specification. The final word purpose is to manage era completely by way of a textual interface, however right here we think about compositional constraints for 2 axes as a proof-of-concept: operate and organism:
Studying the function-structure-sequence connection. PLAID learns the tetrahedral cysteine-Fe2+/Fe3+ coordination sample typically present in metalloproteins, whereas sustaining excessive sequence-level variety.
Coaching utilizing sequence-only coaching information
One other vital side of the PLAID mannequin is that we solely require sequences to coach the generative mannequin! Generative fashions be taught the information distribution outlined by its coaching information, and sequence databases are significantly bigger than structural ones, since sequences are less expensive to acquire than experimental construction.
Studying from a bigger and broader database. The price of acquiring protein sequences is way decrease than experimentally characterizing construction, and sequence databases are 2-4 orders of magnitude bigger than structural ones.
How does it work?
The rationale that we’re capable of prepare the generative mannequin to generate construction by solely utilizing sequence information is by studying a diffusion mannequin over the latent house of a protein folding mannequin. Then, throughout inference, after sampling from this latent house of legitimate proteins, we are able to take frozen weights from the protein folding mannequin to decode construction. Right here, we use ESMFold, a successor to the AlphaFold2 mannequin which replaces a retrieval step with a protein language mannequin.
Our methodology. Throughout coaching, solely sequences are wanted to acquire the embedding; throughout inference, we are able to decode sequence and construction from the sampled embedding. ❄️ denotes frozen weights.
On this means, we are able to use structural understanding info within the weights of pretrained protein folding fashions for the protein design activity. That is analogous to how vision-language-action (VLA) fashions in robotics make use of priors contained in vision-language fashions (VLMs) educated on internet-scale information to produce notion and reasoning and understanding info.
Compressing the latent house of protein folding fashions
A small wrinkle with straight making use of this methodology is that the latent house of ESMFold – certainly, the latent house of many transformer-based fashions – requires quite a lot of regularization. This house can be very massive, so studying this embedding finally ends up mapping to high-resolution picture synthesis.
To deal with this, we additionally suggest CHEAP (Compressed Hourglass Embedding Diversifications of Proteins), the place we be taught a compression mannequin for the joint embedding of protein sequence and construction.
Investigating the latent house. (A) After we visualize the imply worth for every channel, some channels exhibit “huge activations”. (B) If we begin analyzing the top-3 activations in comparison with the median worth (grey), we discover that this occurs over many layers. (C) Huge activations have additionally been noticed for different transformer-based fashions.
We discover that this latent house is definitely extremely compressible. By doing a little bit of mechanistic interpretability to higher perceive the bottom mannequin that we’re working with, we have been capable of create an all-atom protein generative mannequin.
What’s subsequent?
Although we study the case of protein sequence and construction era on this work, we are able to adapt this methodology to carry out multi-modal era for any modalities the place there’s a predictor from a extra plentiful modality to a much less plentiful one. As sequence-to-structure predictors for proteins are starting to sort out more and more complicated programs (e.g. AlphaFold3 can be capable of predict proteins in complicated with nucleic acids and molecular ligands), it’s simple to think about performing multimodal era over extra complicated programs utilizing the identical methodology.
If you’re focused on collaborating to increase our methodology, or to check our methodology within the wet-lab, please attain out!
Additional hyperlinks
For those who’ve discovered our papers helpful in your analysis, please think about using the next BibTeX for PLAID and CHEAP:
@article{lu2024generating,
title={Producing All-Atom Protein Construction from Sequence-Solely Coaching Information},
writer={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Steady Embedding Compressions of Protein Sequence and Construction},
writer={Lu, Amy X and Yan, Wilson and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
You can even checkout our preprints (PLAID, CHEAP) and codebases (PLAID, CHEAP).
Some bonus protein era enjoyable!
Further function-prompted generations with PLAID.
Unconditional era with PLAID.
Transmembrane proteins have hydrophobic residues on the core, the place it’s embedded inside the fatty acid layer. These are constantly noticed when prompting PLAID with transmembrane protein key phrases.
Further examples of lively web site recapitulation based mostly on operate key phrase prompting.
Evaluating samples between PLAID and all-atom baselines. PLAID samples have higher variety and captures the beta-strand sample that has been tougher for protein generative fashions to be taught.
Acknowledgements
Due to Nathan Frey for detailed suggestions on this text, and to co-authors throughout BAIR, Genentech, Microsoft Analysis, and New York College: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin Okay. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, and Nathan C. Frey.
















{kind=link}